Research Studies
Study Tracker
Disruption of the SIRT1/PI3K/AKT Signaling Axis Mediates Fluoride-Induced Cardiotoxicity: Evidence from in Vitro and Zebrafish Models.Abstract
Fluoride (F), an environmental contaminant, is known to induce cardiotoxicity, although the precise molecular mechanisms remain unclear. This study aimed to investigate the role of the SIRT1/PI3K/AKT signaling pathway in F-induced cardiotoxicity and explore the potential protective effects of SIRT1 activation. Human AC16 cardiomyocytes and zebrafish embryos were exposed to increasing concentrations of sodium NaF. Cellular assays were used to assess viability, apoptosis, cell cycle distribution, and oxidative stress. Expression levels of oxidative and inflammatory markers, as well as components of the SIRT1/PI3K/AKT pathway, were analyzed by western blotting, immunofluorescence, and real-time PCR. Zebrafish were evaluated for cardiac developmental abnormalities, apoptosis, and oxidative stress. The SIRT1 agonist SRT1720 was used to evaluate the protective effects of SIRT1 activation. Statistical analysis was performed using SPSS 23 Software and GraphPad Prism7 software, with significant differences evaluated by one-way analysis of variance (ANOVA) and Dunnett’s test (p<0.05). NaF exposure significantly inhibited AC16 cell proliferation, induced G1 phase arrest, and increased apoptosis in a dose-dependent manner. Reactive oxygen species levels were elevated, accompanied by downregulation of antioxidant proteins and upregulation of inflammatory cytokines. NaF markedly suppressed SIRT1, PI3K, and AKT expression while activating FOXO1a. Zebrafish embryos exhibited dose-dependent cardiac malformations, increased apoptosis, and elevated oxidative stress markers. Treatment with SRT1720 restored SIRT1/PI3K/AKT pathway activity, enhanced cell proliferation, reduced apoptosis, and alleviated oxidative and inflammatory responses in both cell and zebrafish models. This study demonstrates that F induces cardiotoxicity by disrupting the SIRT1/PI3K/AKT signaling pathway, leading to increased oxidative stress, inflammation, and apoptosis. Activation of SIRT1 by SRT1720 mitigates these effects, highlighting the protective role of this pathway in F-related cardiac injury. These findings provide mechanistic insights and identify potential molecular targets for the prevention and treatment of fluorosis-associated cardiovascular toxicity.
Introduction
F is widely distributed in the environment in both inorganic and organic forms and plays an important role in the growth and development of humans and animals [1, 2]. While moderate F intake is beneficial, particularly in preventing dental caries, excessive exposure has been associated with adverse health effects [3]. Research has shown that chronic overexposure to F can lead to toxic damage in multiple organs, including the brain, heart, kidneys, liver, and reproductive system [4,5,6]. Among these, the heart, a vital organ responsible for maintaining circulatory function, is particularly susceptible to F-induced toxicity. Animal studies have demonstrated significant histopathological and biochemical alterations in myocardial tissue following prolonged sodium F exposure, including myocardial cell necrosis, cytoplasmic vacuolization, myocyte nuclear degradation, and degenerative changes in myocardial fibers [7]. An epidemiological study of 26 patients with fluorosis pointed out that excessive intake of F can lead to a decrease in myocardial contractility [8]. In addition, excessive F intake is closely related to changes in human electrocardiograms, cardiac hypertrophy, peripheral vascular diseases, ventricular diastolic dysfunction, and carotid atherosclerosis [9, 10]. The above research results show that excessive intake of F can cause structural and functional defects in the heart, eventually leading to a series of cardiovascular diseases. However, the specific mechanism by which F causes cardiovascular diseases in humans still needs further exploration.
Fluorosis is closely associated with oxidative stress. F ions can disrupt the mitochondrial electron transport chain, leading to excessive production of reactive oxygen species (ROS), including superoxide anions (O2–) and hydrogen peroxide (H2O2) [11]. This overproduction of ROS induces mitochondrial oxidative stress, which in turn impairs mitochondrial biogenesis, disrupts fatty acid metabolism, and compromises antioxidant defense systems. Recent studies have established a strong link between mitochondrial dysfunction and the pathogenesis of several cardiovascular diseases, such as myocardial infarction, cardiac hypertrophy, and heart failure [12]. Consequently, strategies aimed at reducing oxidative stress or inhibiting apoptosis may offer promising molecular targets for mitigating F-induced cardiotoxicity. The PI3K/AKT signaling pathway plays a central role in cardioprotection, with functions spanning cell survival, myocardial contractility, electrophysiological regulation, hypertrophic remodeling, and energy metabolism [13]. As a downstream effector of phosphoinositide 3-kinase (PI3K), AKT helps preserve mitochondrial function and prevent apoptosis by modulating BCL-2 family proteins and caspase activation, particularly caspase-8 and caspase-3 [14]. In parallel, sirtuin 1 (SIRT1), a NAD+-dependent deacetylase, is widely recognized for its regulatory role in oxidative stress, inflammation, mitochondrial function, autophagy, and programmed cell death. SIRT1 modulates the expression of brain-derived neurotrophic factor (BDNF) through the deacetylation of transcription factors and histones. BDNF binding to its receptor TrkB subsequently activates FOXO and PI3K signaling, influencing ROS generation and inflammatory responses [15,16,17,18,19].
Despite the established roles of oxidative stress, the PI3K/AKT pathway, and SIRT1 in cardiac health, a comprehensive understanding of whether F-induced cardiotoxicity is mediated through the ROS-dependent dysregulation of the SIRT1/PI3K/AKT signaling axis remains elusive. Addressing this knowledge gap is crucial for identifying novel therapeutic targets to alleviate F-related cardiac injury. This study aimed to investigate the cardiotoxic effects of sodium NaF and elucidate the role of the SIRT1/PI3K/AKT signaling axis in mediating oxidative stress, inflammation, and apoptosis. Using both human AC16 cardiomyocytes and zebrafish as complementary experimental models, we provide mechanistic insights into how F disrupts cardiac cellular homeostasis and evaluate the therapeutic potential of SIRT1 activation in mitigating its toxic effects.
Methods
Preparation of NaF and SRT1720
NaF was obtained from Jinshan City Co., Ltd. (Chengdu, China) and dissolved in purified water to prepare a 10 mM stock solution. The solution was sterilized by filtration through a 0.22 um membrane filter, aliquoted, and stored at 4 °C until use. The SIRT1 agonist SRT1720 (MedChemExpress, Monmouth Junction, NJ, USA,) was similarly diluted in purified water to 50 uM and stored at 4 °C. 2 uM SRT1720 was selected as the intervention concentration in this study [20].
AC16 Cell Culture
Human AC16 cardiomyocytes were provided from Fuheng Biology (Shanghai, China) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (GIBCO, Grand Island, NY, USA) and 100 ug/mL penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). Cells were maintained at 37 °C in a humidified incubator with 5% CO2.
Zebrafish Maintenance and Embryo Exposure
Zebrafish are widely used as the main ecotoxicological model in the field of life science research due to their advantages such as small size, high reproductive performance, rapid organ development, similar morphology and physiology to mammals, and high sensitivity to harmful substances [21]. Therefore, in this study, zebrafish were used to clarify the cardiotoxicity and oxidative stress induced by sodium F, as well as the interaction relationship of the SIRT1/PI3K/AKT signaling axis.
Adult wild-type AB strain zebrafish male and female were obtained from the China Zebrafish Resource Center and maintained under standard conditions (14 h light/10 h dark cycle, 28 °C, pH 7.5–8.0, conductivity 980–1000 uS/cm). The fish were fed brine worms twice daily. Breeding pairs were placed in isolated spawning boxes (Tecniplast, Varese, Italy) overnight to prevent egg predation. The following morning, the dividers were removed, and light exposure initiated the spawning cycle. Zebrafish embryos and larvae were subsequently cultured in a non-CO2 incubator under identical photoperiod and temperature conditions. To determine appropriate NaF exposure concentrations, we referred to previous studies reporting the 50% lethal concentration (LC50) of NaF for zebrafish as 0.92 mM [22]. Based on this, four treatment concentrations (0, 0.25, 0.5, and 1.0 mM NaF) were selected for the experiments.
Cell Viability Assay (CCK-8)
AC16 cells (5×103 cells/well) were seeded in 96-well plates and incubated overnight. Cells were then treated with NaF at 0, 0.0625, 0.125, 0.25, 0.5, 1.0, or 2.0 mM for 24 h. Cell viability was assessed using a CCK-8 kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Absorbance was measured at 590 nm using a microplate reader (Thermo Fisher Scientific), and proliferation was expressed as a percentage relative to control.
Crystal Violet Staining
AC16 cells were inoculated into 12-well cell culture plates at a density of 2×105 cells/mL and cultured at 37 °C with 5% CO2 for 24 h. Upon reaching 50% confluence, cells were treated with 0, 0.25, 0.5, or 1.0 mM NaF for an additional 24 h. For subsequent intervention experiments, the control group received 0 mM NaF, the NaF group received 0.5 mM NaF, and the SRT1720 group received a combination of 0.5 mM NaF and 2 uM SRT1720. Cells were then fixed with 4% paraformaldehyde, rinsed twice with phosphate-buffered saline (PBS), and stained with a 1% crystal violet solution (Solarbio, Beijing, China). Cell morphological changes were observed, analyzed, and recorded using an inverted microscope (Leica DM2500, Heidelberg, Germany).
5-Ethynyl-2’-Deoxyuridine (EdU) Assay
AC16 cells in the logarithmic growth phase were seeded into 24-well plates at a density of 2×105 cells per well and cultured for 24 h. Following this, cells were exposed to various concentrations of NaF (0, 0.25, 0.5, 1.0 mM) and cultured for another 24 h. For intervention experiments, the control group received 0 mM NaF, the NaF group received 0.5 mM NaF, and the SRT1720 group received a combination of 0.5 mM NaF and 2 uM SRT1720. An EdU solution, diluted at a 1:1000 ratio, was then added, and cells were cultured for an additional 2 h. Subsequent steps adhered strictly to the manufacturer’s guidelines. Cells were observed using an inverted fluorescence microscope. Three random fields of view were captured from each well, and the fluorescence intensity of EdU-positive cells was quantified using ImageJ 1.8.0 software (NIH, Bethesda, MD, USA). The percentage of EdU-positive cells was calculated as: (red EdU-positive nuclei/blue DAPI-stained nuclei)×100%.
Intracellular ROS Measurement
AC16 cells were inoculated in 12-well plates at a density of 2×105 cells/mL and incubated for 24 h. Cells were then exposed to 0, 0.25, 0.5, and 1.0 mM NaF for 24 h. Intracellular ROS levels were detected using the reactive oxygen species assay kit (Jiancheng Bioengineering, Nanjing, China). Staining was performed with 2,7-dichlorofluorescein diacetate (DCFH-DA, Molecular Probes, Eugene, OR, USA) and observed using an inverted microscope. To evaluate ROS distribution in zebrafish, embryos were exposed to 0, 0.25, 0.5, and 1.0 mM NaF for 24 h. Embryos were then rinsed twice with embryo culture medium. A DCFH-DA probe, with a final concentration of 30 uM, was added, and embryos were incubated in the dark at 280C for 1 h. After staining, zebrafish were imaged and observed using an inverted fluorescence microscope.
Flow Cytometry for Apoptosis and Cell Cycle
Cells were seeded into 6-well plates at a density of 3×106 cells/mL and incubated for 24 h. Cells were subsequently exposed to NaF solutions at concentrations of 0, 0.25, 0.5, and 1.0 mM and incubated for an additional 24 h. For intervention experiments, the control group received 0 mM NaF, the NaF group received 0.5 mM NaF, and the SRT1720 group received a combination of 0.5 mM NaF and 2 uM SRT1720. Flow cytometry for apoptosis detection was performed using the fluorescein isothiocyanate (FITC)-labeled Annexin V/PI apoptosis detection kit (KEYGEN, Nanjing, China), following the manufacturer’s protocol. For cell cycle detection, collected cells were immobilized in cold ethanol and stored overnight at 4oC. The following day, cells were washed twice with cold PBS. According to the instructions of the Cell Cycle Kit (KEYGEN), 100 uL of RNase A (25 ug/mL) and 400 uL of PI (50 ug/mL) were added to each sample. Samples were then incubated in the dark for 30 min before analysis by flow cytometry.
Immunofluorescence for SIRT1
AC16 cells were inoculated into 12-well plates at a density of 2×105 cells per well. The next day, cells were continuously exposed to NaF solutions at concentrations of 0, 0.25, 0.5, and 1.0 mM for 24 h. Subsequently, cells were fixed and permeabilized, then blocked with goat serum. Cells were incubated overnight at 4oC with a SIRT1-specific antibody (diluted at 1:200; Proteintech, Rosemont, IL, USA), followed by incubation with a secondary antibody conjugated with SIRT1 for 2 h. Finally, fluorescence images were captured using confocal microscopy. Fluorescence intensities were calculated as: (Fluorescence intensity of the experimental group/(fluorescence intensity of the control group)×100%.
Cardiac Morphology in Zebrafish
Zebrafish treated with 0, 0.25, 0.5, and 1.0 mM NaF were selected at 120 h post-fertilization (hpf). They were anesthetized with 150 mg/L tricaine and fixed in 3% methylcellulose (Sigma St. Louis, MO, USA) to capture side-view images. The morphology of the heart was observed and photographed using a ZEISS Discovery.V20 microscope (Oberkochen, Germany) equipped with a Nikon camera (Tokyo, Japan).
Acridine Orange (AO) Staining for Apoptosis in Zebrafish
Apoptotic signals in the hearts of zebrafish larvae were detected using AO staining. After 96 h of exposure, 15 juvenile fish were randomly selected from each concentration group and anesthetized. They were then washed three times with PBS buffer solution, 5 min each wash. Subsequently, juvenile fish were rinsed three times with a 30% (v/v) Danieau’s solution (58 mM NaCl, 0.7 mM KCl, 0.4 mM MgSO4, 0.6 mM Ca(NO3)2 and 5 mM HEPES, pH 7.4). Finally, the juvenile fish were transferred to a 30% Danieau’s solution containing 5 ug/mL AO and incubated for 20 min. Apoptosis was assessed by green fluorescence in the heart region.
Western Blotting
Total protein was extracted from AC16 cells or zebrafish tissues using RIPA buffer with protease inhibitors. Protein concentration was determined, and equal amounts were subjected to SDS-PAGE and transferred to PVDF membranes. Membranes were blocked, incubated overnight with primary antibodies (targeting HO-1, NQO1, NRF2, IL-1, IL-6, Caspase-3/8, BAX, BCL-2, SIRT1, PI3K, AKT, FOXO1a, etc.), followed by HRP-conjugated secondary antibody incubation and ECL detection. Protein band intensity was quantified using ImageJ and normalized to B-actin.
Real-Time Fluorescence Quantitative PCR (RT-qPCR)
Total RNA from AC16 cells was extracted using Trizol reagent (Solarbio, Beijing, China) and 1000 ng of RNA from each sample was reverse-transcribed using EasyScript® SuperMix (TransGen Biotech, Beijing, China). Primer sequences are listed in Table 1. qPCR was performed using PerfectStart® Green qPCR SuperMix (TransGen Biotech) on triplicate samples. The expression level of each gene was normalized to B-actin using the 2???Ct method.
Statistical Analysis
All data are repeated three times or more. Results are presented as standard error of the mean (mean±SD). All data analyses were conducted using SPSS 23 Software (IBM Corp., Armonk, NY, USA) and GraphPad Prism 7 software (GraphPad Software Inc., La Jolla, CA, USA). Significant differences between the exposure group and the control group were evaluated through one-way analysis of variance (ANOVA), with the Dunnett test performed using SPSS 23. A p-value<0.05 was considered statistically significant.
Results
NaF Inhibits AC16 Cell Viability and Proliferation
Cell viability was assessed using the CCK-8 assay. Exposure to increasing concentrations of NaF significantly inhibited AC16 cell proliferation in a dose-dependent manner (p<0.001; Fig. 1A). The half-maximal inhibitory concentration (LD50) was determined to be 0.45±0.03 mM (Fig. 1B). Based on this result, NaF concentrations of 0.25, 0.5, and 1.0 mM were selected for subsequent experiments. Crystal violet staining confirmed a concentration-dependent reduction in cell number and morphological changes, including cell rounding, shrinkage, and detachment (Fig. 1C). EdU fluorescence staining further demonstrated a significant dose-dependent decrease in the number of proliferating (EdU-positive) AC16 cells after NaF exposure (Fig. 1D).
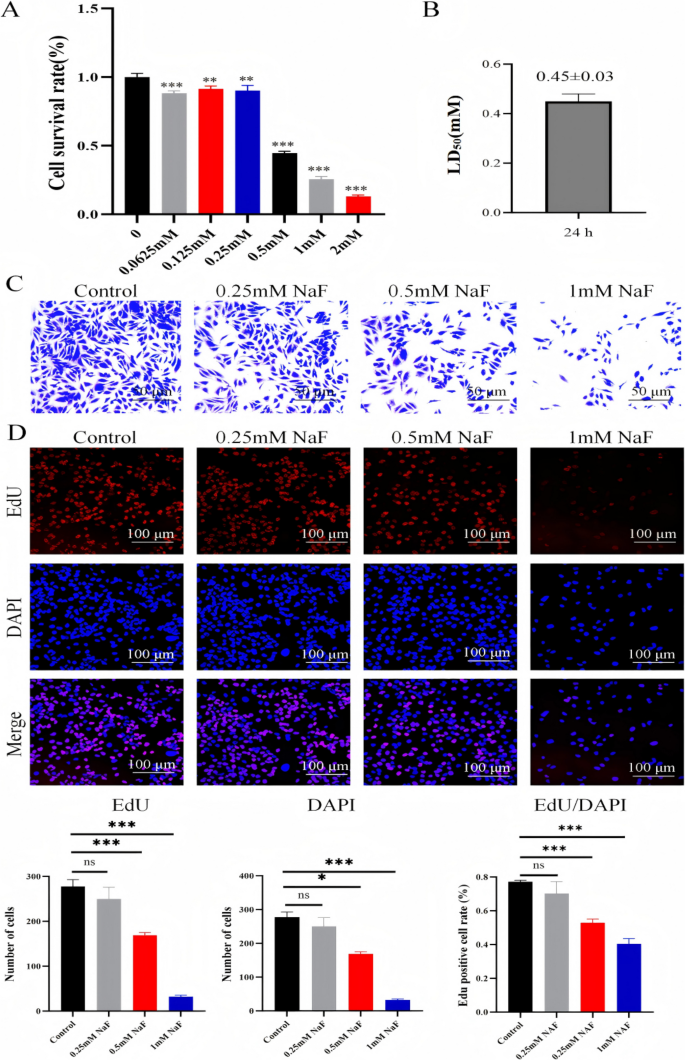
Effects of NaF on the proliferation of AC16 cells. A CCK-8 assay of AC16 cells treated with 0, 0.0625, 0.125, 0.25, 0.5, 1, and 2.0 mM NaF for 24 h. B LD50 of AC16 cells following 24-h NaF exposure. C Morphological changes in AC16 cells after treatment with 0, 0.25, 0.5, and 1.0 mM NaF for 24 h, observed by optical microscopy (× 200). D EdU assay showing cell proliferation after NaF treatment at indicated concentrations for 24 h (×100, n=3). Values are expressed as mean SD. *p<0.05, **p<0.01, ***p<0.001 vs. control
NaF Induces Apoptosis in AC16 Cells and Disrupts Cell Cycle Progression
Flow cytometry revealed that NaF significantly increased both early and late apoptosis in AC16 cells in a concentration-dependent manner (p<0.001; Fig. 2A). Western blotting analysis showed upregulation of pro-apoptotic proteins caspase-3, caspase-8, p-P53, BAX, and P21 (all p<0.01), while anti-apoptotic BCL-2 expression was significantly decreased (p<0.01; Fig. 2B). NaF treatment caused a significant accumulation of AC16 cells in the G1 phase and a corresponding reduction in the S phase, indicating cell cycle arrest (Fig. 2C). Western blotting further confirmed a concentration-dependent decrease in the expression of CDK2 (p<0.01), C-MYC (p<0.001), and Cyclin D1 (p<0.001) proteins (Fig. 2D).
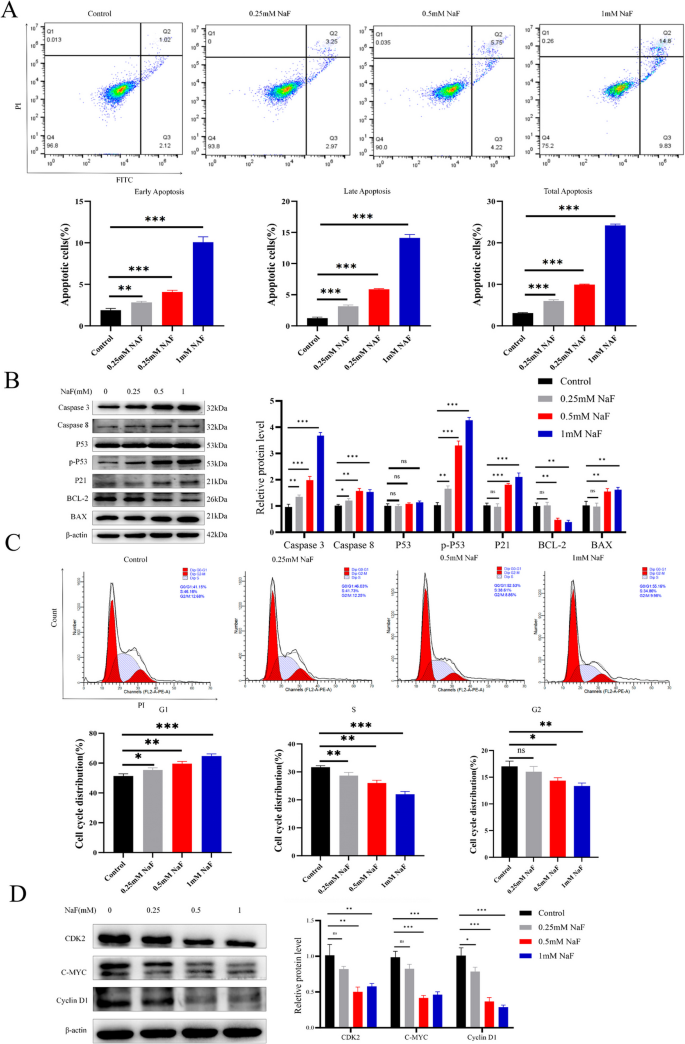
NaF significantly promotes apoptosis and disrupts the cell cycle in AC16 cells. A Flow cytometry showing NaF-induced early and late apoptosisn(n=3). B NaF dose-dependently downregulated BCL-2 and upregulated Caspase-3, Caspase-8, p-P53, BAX, and P21 protein levels (n=3). C Flow cytometry analysis showing G1 phase arrest after NaF treatment (n=3). D Protein expression of CDK2, C-MYC, and Cyclin D1 decreased with increasing NaF concentrations (n=3). Values are expressed as mean±SD. *p<0.05, **p<0.01, ***p<0.001 vs. control
NaF Elevates ROS and Inflammatory Markers in AC16 Cells, Suppresses the SIRT1/PI3K/AKT Pathway and Activates FOXO1a
Intracellular ROS levels increased significantly with rising NaF concentrations (p<0.001; Fig. 3A). Consistent with oxidative stress induction, western blotting showed downregulation of antioxidant proteins HO-1, NQO1 and NRF2 (both p<0.01), and upregulation of inflammatory cytokines IL-1 and IL-6 (both p<0.001; Fig. 3B). Immunofluorescence staining showed that NaF significantly reduced SIRT1 expression in AC16 cells (p<0.001; Fig. 3C). Western blotting results further revealed that NaF downregulated protein levels of SIRT1, PI3K, and AKT (all p<0.001), while upregulating FOXO1a expression (p<0.001; Fig. 3D), indicating dysregulation of the SIRT1/PI3K/AKT axis.
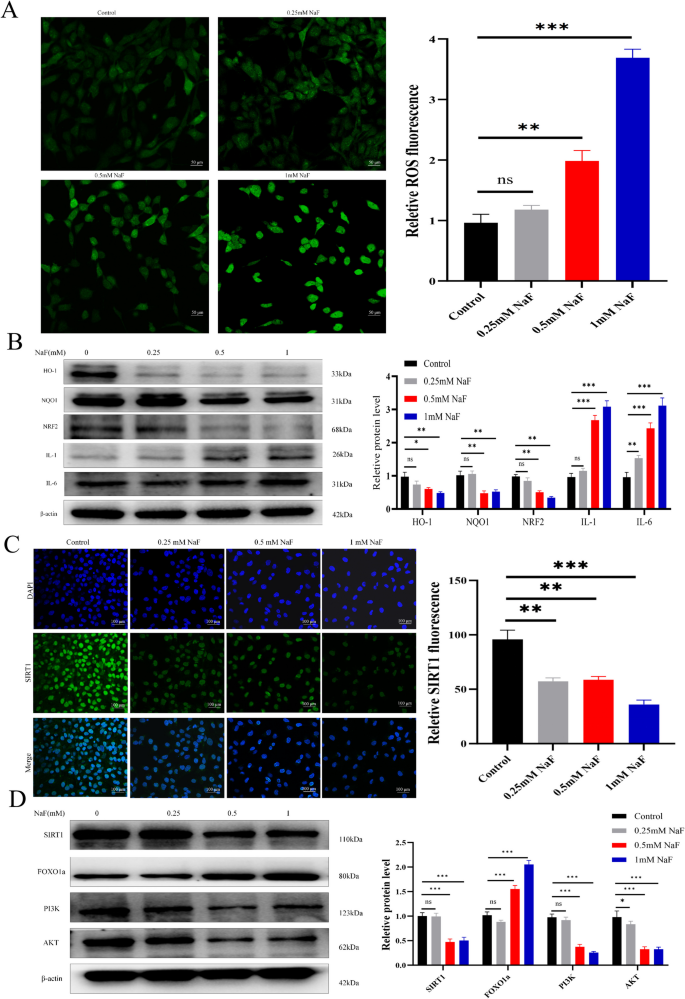
NaF induces oxidative stress and inflammatory responses, modulates the SIRT1 pathway and its downstream targets in AC16 cells. A ROS levels in AC16 cells after 24-h NaF treatment (×200, n=3). B NaF decreased HO-1, NQO1 and NRF2, while increasing IL-1 and IL-6 expression (n=3). C Immunofluorescence shows decreased SIRT1 expression with increasing NaF concentration (×100, n=3). D Western blotting results show upregulation of FOXO1a and downregulation of SIRT1, PI3K, and AKT with higher NaF concentrations (n=3). Values are expressed as mean±SD. *p<0.05, **p<0.01, ***p<0.001 vs. control
NaF Induces Cardiac Abnormalities in Zebrafish Embryos and Induces Apoptosis in Zebrafish Heart Tissue
During zebrafish embryogenesis, the heart first appears as a linear tube that undergoes a tightly regulated morphogenetic sequence, known as cardiac looping, in which it bends, rotates, and partitions to form distinct atrial, ventricular, and atrioventricular structures. NaF exposure disrupted normal cardiac looping in zebrafish embryos. Increasing NaF concentrations resulted in progressive defects in cardiac morphogenesis, including failure of cardiac tube bending, chamber separation, and proper ventricular formation (Fig. 4A). Acridine orange staining revealed strong green fluorescence in the hearts of zebrafish larvae exposed to NaF (0.25–1.0 mM), indicating apoptosis, while no signal was observed in control larvae (Fig. 4B). RT-qPCR analysis further showed that compared to the control group, the expression levels of apoptosis-related genes caspase-3, caspase-8, and BAX significantly increased in the hearts of zebrafish larvae at each NaF concentration (p<0.001 or p<0.01). Conversely, the expression of BCL-2 was significantly decreased (p<0.001) (Fig. 4C).
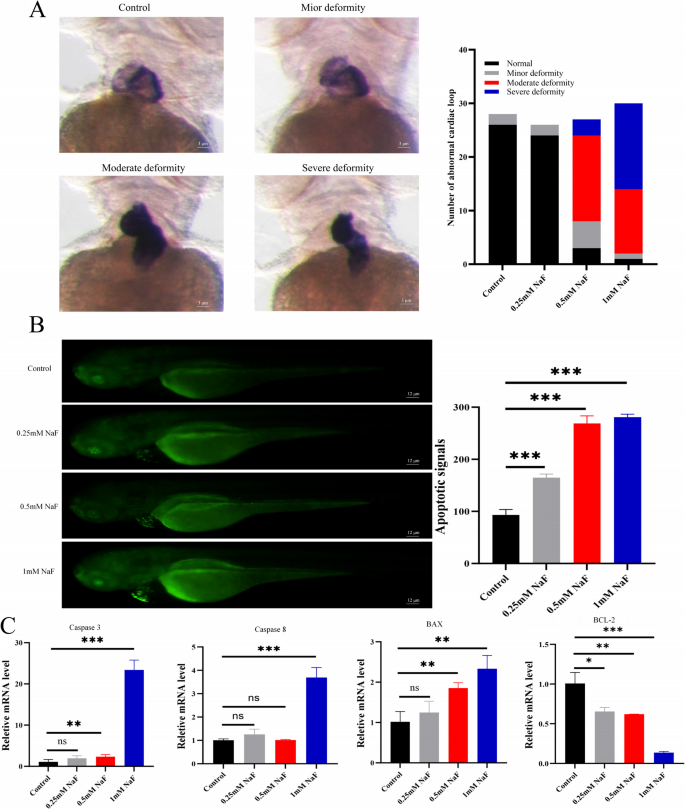
Abnormal cardiac morphology in zebrafish after NaF exposure. A Confocal microscopy (× 1600) showing defects in cardiac looping with increasing NaF concentrations and quantification of the proportion of zebrafish with abnormal cardiac morphology across treatment groups (n=3). B AO staining (×400) showing apoptotic signals in zebrafish larvae after NaF exposure (n=3). C qPCR results indicate a dose-dependent decrease in BCL-2 and increase in Caspase-3, Caspase-8, and BAX mRNA expression (n=20). Values are expressed as mean±SD. *p<0.05, **p<0.01, ***p<0.001 vs. control
NaF Promotes ROS Generation and Inflammation in Zebrafish
ROS levels in zebrafish models were assessed at both 72 and 96 h post-fertilization (hpf), with ROS staining performed 24 h after NaF treatment. Experimental results demonstrated a significant upward trend in ROS levels with increasing NaF treatment dose compared to the control group (Fig. 5A-B). In parallel, qPCR showed reduced expression of HO-1 and NQO1 (both p<0.001) and increased IL-1 expression (p<0.001; Fig. 5C), consistent with oxidative stress and inflammatory responses.
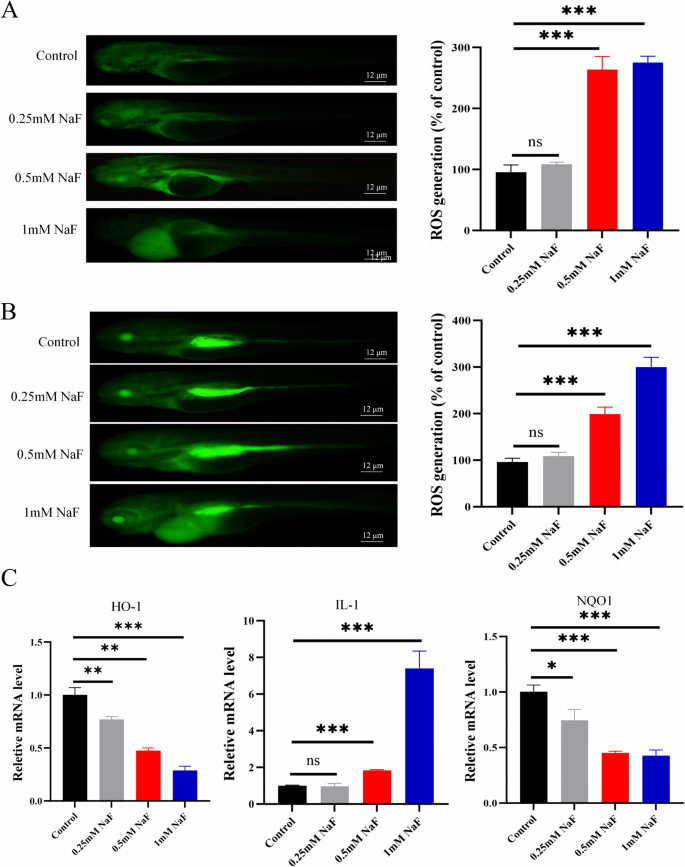
NaF promotes oxidative stress and inflammation in zebrafish. A-B ROS detection (×400) after 24-h NaF treatment (n=3). C NaF exposure downregulated Nqo-1 and Ho-1 and upregulated IL-1 expression (n=20). Values are expressed as mean±SD. *p<0.05, **p<0.01, ***p<0.001 vs. control
SRT1720 Mitigates NaF-Induced Cytotoxicity in AC16 Cells
Crystal violet staining results demonstrated that compared to the NaF group, the combined NaF and SRT1720 treatment significantly restored cell morphology and increased cell numbers (Fig. 6A). Further EdU assays confirmed that the combined NaF and SRT1720 treatment group exhibited a significant increase in the number of positive cells, total cell count, and the percentage of EdU-positive cells (p<0.05) when compared to the NaF-only group (Fig. 6B). These findings indicate that SRT1720 effectively alleviates the inhibitory effect of NaF on the proliferative capacity of AC16 cells.

SRT1720 mitigates NaF-induced toxicity in AC16 cells. A Cell morphology in control, NaF, and NaF+SRT1720 groups (×200). B EdU assay showing proliferation recovery in the SRT1720-treated group (×200, n=3). Values are expressed as mean±SD. *p<0.05, **p<0.01, ***p<0.001 vs. NaF group
SRT1720 Reduces Apoptosis and Restores Cell Cycle in NaF-Treated Cells
Compared to the NaF group, flow cytometry showed that SRT1720 significantly decreased early, late, and total apoptosis in NaF-exposed AC16 cells (p<0.001; Fig. 7A). It also reversed NaF-induced cell cycle arrest, increasing S-phase and reducing G1-phase populations (p<0.001; Fig. 7B).
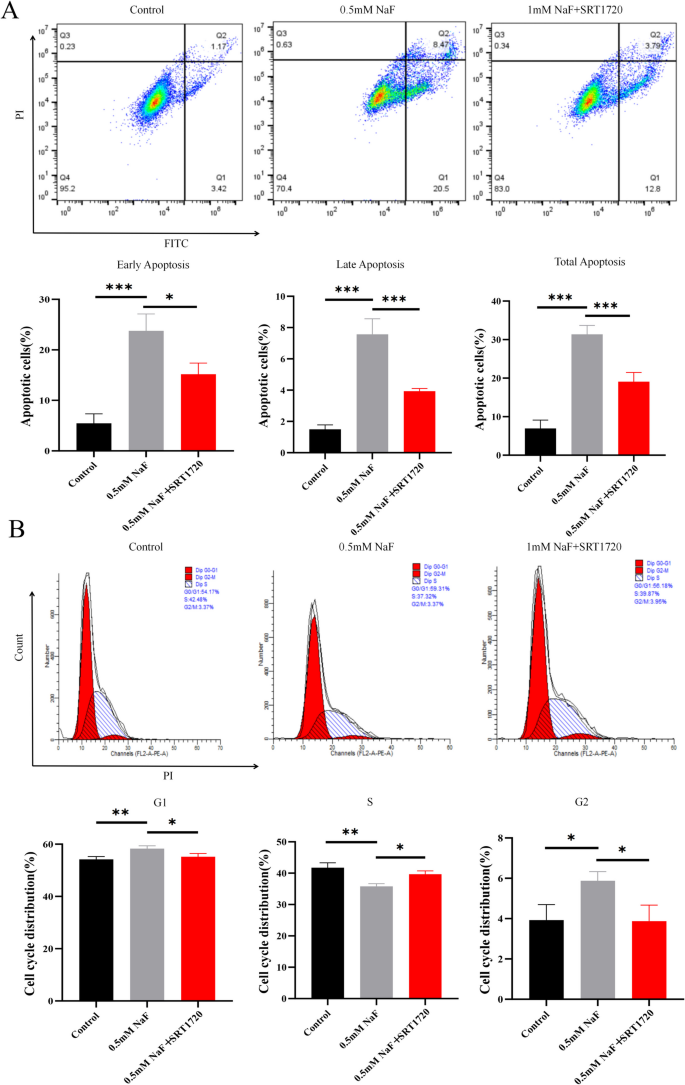
SRT1720 alleviates apoptosis and restores the cell cycle in NaF-treated AC16 cells. A Flow cytometry showing reduced apoptosis following SRT1720 treatment (n=3). B Cell cycle analysis indicating recovery of progression with SRT1720 (n=3). Values are expressed as mean±SD. *p<0.05, **p<0.01, ***p<0.001 vs. NaF group
SRT1720 Activates the Expression of PI3K/AKT Pathway Proteins
Compared with the control group, NaF significantly downregulated the protein levels of SIRT1 (p<0.01), PI3K (p<0.01), and AKT (p<0.05), while upregulating the expression of FOXO1a (p<0.01) (Fig. 8A). In contrast, treatment with SRT1720 in the presence of NaF significantly increased the protein levels of SIRT1, PI3K, and AKT (all p<0.05), and simultaneously reduced FOXO1a expression (p<0.05) compared with the NaF group (Fig. 8A). The flow chart of the study is shown in Fig. 8B.
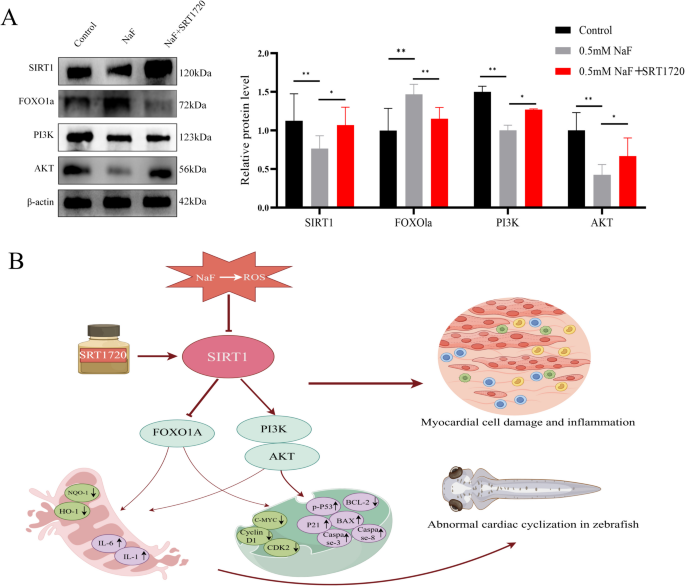
SIRT1 activation by SRT1720 reduces NaF-induced damage in AC16 cells. A SRT1720 upregulated SIRT1, PI3K, and AKT, while downregulating FOXO1a (n=3). B Schematic diagram illustrating the proposed mechanism of this study. Values are expressed as mean±SD. *p<0.05, **p<0.01, ***p<0.001 vs. NaF group
Discussion
Fluorosis has emerged as a significant global public health concern. Epidemiological studies have shown that both the incidence and mortality rates of heart disease are markedly higher among individuals residing in fluorosis-endemic areas compared to those in non-affected regions [23]. A growing body of evidence has demonstrated the detrimental impact of fluorosis on cardiac health, including reduced cardiac output, the development of arrhythmias, and disturbances in cardiac conduction such as blockages. Chronic excessive F intake can impair both the structural integrity and functional capacity of the heart, potentially leading to serious cardiovascular complications.
The heart is a critical non-skeletal target organ for F toxicity. Increasing exposure to NaF has been shown to cause myocardial tissue congestion and damage to muscle fibers, ultimately impairing myocardial contractile function [24]. Numerous studies have further demonstrated that NaF reduces cell viability, inhibits DNA synthesis, and induces G1 phase cell cycle arrest [25]. In the present study, a dose-dependent decline in AC16 cell proliferation was observed with increasing NaF concentrations, indicating a strong negative correlation between NaF exposure and cellular growth. NaF disrupted the cell cycle progression of AC16 cells, with the degree of impairment intensifying alongside higher concentrations. Morphological observations revealed that NaF-treated AC16 cells became curled, contracted, and detached from the culture surface. Consistently, in zebrafish embryos, the severity of abnormal cardiac looping increased with rising NaF concentrations, further emphasizing the cardiotoxic potential of F exposure.
Studies have shown that in soft tissues, NaF interferes with cellular function by inhibiting multiple enzymes, ultimately leading to the generation of free radicals [26]. Its accumulation compromises the body’s ability to eliminate these radicals, which can severely damage biological membranes. An imbalance between increased free radical production and impaired antioxidant defenses results in oxidative stress [27]. Consistent with these findings, both our in vitro AC16 cell model and in vivo zebrafish experiments demonstrated that NaF exposure elevated oxidative stress in cardiomyocytes in a concentration-dependent manner. Concurrently, the expression levels of pro-inflammatory cytokines IL-1 and IL-6 increased, while antioxidant proteins NQO-1 and HO-1 were downregulated. These results suggest that F exposure can induce oxidative stress and activate inflammatory pathways in myocardial cells, thereby contributing to cardiac injury. Maintaining antioxidant homeostasis is therefore essential for preserving normal cardiac function. Numerous studies have identified caspase activation as a key molecular event in apoptosis [28, 29], with caspase-8 functioning as an initiator of the mitochondrial apoptotic pathway and caspase-3 serving as a central executioner [30]. Our results showed that increasing NaF concentrations upregulated both caspase-3 and caspase-8, which correlated with the extent of cardiomyocyte apoptosis. These findings highlight the importance of elucidating the underlying mechanisms by which NaF induces cardiotoxicity.
Studies have demonstrated that SIRT1 plays a pivotal role in cardiac developmental toxicity induced by trace elements such as fluorine [31]. SIRT1 is a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase that contributes to the maintenance of cardiac homeostasis [32]. It regulates key cellular processes, including differentiation, autophagy, apoptosis, inflammatory responses, energy metabolism, and oxidative stress, through the activation of various signaling pathways [33, 34]. Enhanced SIRT1 activity confers resistance to oxidative stress by modulating the activity of FOXO transcription factors [35]. SIRT1 directly interacts with FOXO1, deacetylating its residues that are otherwise acetylated by cAMP response element-binding proteins (CREB) [36]. Overexpression of SIRT1 has been shown to protect cardiomyocytes from oxidative damage via a FOXO1-dependent mechanism [37]. In parallel, F-induced ROS production and apoptosis have been linked to dysregulation of the PI3K/AKT signaling pathway [38]. For example, baicalin has been shown to mitigate oxidative stress, inflammation, and apoptosis by activating the PI3K/AKT pathway [39]. Consistent with these findings, our study demonstrated that NaF exposure inhibited SIRT1 expression and altered the levels of FOXO1a, PI3K, and AKT proteins, activating the FOXO1a pathway while suppressing PI3K/AKT signaling. Importantly, the use of SRT1720 to activate SIRT1 in NaF-treated AC16 cells restored cell proliferation and reduced apoptosis. This was accompanied by partial recovery of SIRT1 signaling, downregulation of FOXO1a expression, and upregulation of PI3K and AKT proteins. These findings support a protective role of SIRT1 in mitigating NaF-induced cardiotoxicity through coordinated regulation of FOXO1a and PI3K/AKT pathways.
Therefore, SIRT1 regulates apoptosis and oxidative stress by modulating the FOXO1 and PI3K/AKT signaling pathways. Through this dual regulation, it reduces oxidative stress and the expression of inflammatory mediators, thereby exerting a significant protective effect against F-induced cardiomyocyte toxicity. By preserving myocardial cell integrity and minimizing tissue damage, SIRT1 emerges as a potential therapeutic target. This discovery provides an important theoretical foundation for the prevention and management of fluorosis-related cardiac injury.
Conclusions
This study provides new evidence that NaF induces cardiotoxicity by promoting oxidative stress, inflammation, and apoptosis in cardiomyocytes. These toxic effects are mediated, at least in part, by dysregulation of the SIRT1/PI3K/AKT signaling axis. NaF suppresses SIRT1 and PI3K/AKT expression while activating FOXO1a, thereby exacerbating cellular injury. Activation of SIRT1 by its agonist SRT1720 significantly attenuated NaF-induced cytotoxicity, restored antioxidant and anti-apoptotic responses, and reactivated the PI3K/AKT pathway, underscoring the cardioprotective role of SIRT1. These findings not only clarify the molecular mechanisms underlying F-induced cardiac injury but also highlight SIRT1 as a promising therapeutic target for mitigating fluorosis-related cardiovascular damage. The study provides a valuable theoretical basis for the development of preventive and interventional strategies against environmental F exposure and its systemic health risks.
Data Availability
No datasets were generated or analysed during the current study.
References
-
Jianjie C, Wenjuan X, Jinling C et al (2016) Fluoride caused thyroid endocrine disruption in male zebrafish (Danio rerio). Aquat Toxicol 171:48–58. https://doi.org/10.1016/j.aquatox.2015.12.010
-
Li M, Cao J, Chen J et al (2016) Waterborne fluoride exposure changed the structure and the expressions of steroidogenic-related genes in gonads of adult zebrafish (Danio rerio). Chemosphere 145:365–375. https://doi.org/10.1016/j.chemosphere.2015.11.041
-
Camargo JA (2003) Fluoride toxicity to aquatic organisms: a review. Chemosphere 50(3):251–264. https://doi.org/10.1016/s0045-6535(02)00498-8
-
Adali MK, Varol E, Aksoy F et al (2013) Impaired heart rate recovery in patients with endemic fluorosis. Biol Trace Elem Res 152(3):310–315. https://doi.org/10.1007/s12011-013-9627-6
-
Perumal E, Paul V, Govindarajan V et al (2013) A brief review on experimental fluorosis. Toxicol Lett 223(2):236–251. https://doi.org/10.1016/j.toxlet.2013.09.005
-
Mukherjee S, Sarkar O, Chattopadhyay A (2025) Individual and combined effects of fluoride and arsenic on gut bacteria: a recent update. Nucleus 68:213–226. https://doi.org/10.1007/s13237-023-00460-4
-
Cicek E, Aydin G, Akdogan M et al (2005) Effects of chronic ingestion of sodium fluoride on myocardium in a second generation of rats. Hum Exp Toxicol 24(2):79–87. https://doi.org/10.1191/0960327105ht505oa
-
Varol E, Akcay S, Ersoy IH et al (2010) Impact of chronic fluorosis on left ventricular diastolic and global functions. Sci Total Environ 408(11):2295–2298. https://doi.org/10.1016/j.scitotenv.2010.02.011
-
Flora SJ, Pachauri V, Mittal M et al (2011) Interactive effect of arsenic and fluoride on cardio-respiratory disorders in male rats: possible role of reactive oxygen species. Biometals 24(4):615–628. https://doi.org/10.1007/s10534-011-9412-y
-
Yan X, Dong N, Hao X et al (2019) Comparative transcriptomics reveals the role of the Toll-like receptor signaling pathway in fluoride-induced cardiotoxicity. J Agric Food Chem 67(17):5033–5042. https://doi.org/10.1021/acs.jafc.9b00312
-
Babu S, Manoharan S, Ottappilakkil H et al (2022) Role of oxidative stress-mediated cell death and signaling pathways in ex-perimental fluorosis. Chem Biol Interact 365:110106. https://doi.org/10.1016/j.cbi.2022.110106
-
Okonko DO, Shah AM (2015) Heart failure: mitochondrial dysfunction and oxidative stress in CHF. Nat Rev Cardiol 12(1):6–8, 2014.189. https://doi.org/10.1038/nrcardio.2014.189
-
Rota M, Boni A, Urbanek K et al (2005) Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ Res 97:1332–1341. https://doi.org/10.1161/01.RES.0000196568.11624.ae
-
Li D, Ni S, Miao KS et al (2019) PI3K/Akt and caspase pathways mediate oxidative stress-induced chondrocyte apoptosis. Cell Stress Chaperones 24(1):195–202. https://doi.org/10.1007/s12192-018-0956-4
-
Hwang JW, Yao H, Caito S et al (2013) Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic Biol Med 61:95–110. https://doi.org/10.1016/j.freeradbiomed.2013.03.015
-
Lu C, Zhao H, Liu Y et al (2023) Novel role of the SIRT1 in endocrine and metabolic diseases. Int J Biol Sci 19(2):484–501. https://doi.org/10.7150/ijbs.78654
-
Patel S, Khan H, Majumdar A et al (2022) Crosstalk between sirtuins and Nrf2: SIRT1 activators as emerging treatment for diabetic neuropathy. Metab Brain Dis 37(7):2181–2195. https://doi.org/10.1007/s11011-022-00956-z
-
Meng T, Qin W, Liu B (2020) SIRT1 Antagonizes Oxidative Stress in Diabetic Vascular Complication. Front Endocrinol (Lausanne) 16(11):568861. https://doi.org/10.3389/fendo.2020.568861
-
Wang J, Song X, Tan G et al (2021) NAD+ improved experimental autoimmune encephalomyelitis by regulating SIRT1 to inhibit PI3K/Akt/mTOR signaling pathway. Aging (Albany NY) 13(24):25931–25943. https://doi.org/10.18632/aging.203781
-
Sancak B, Inönü D, Alp G et al (2025) Cardioprotective role of SIRT1 activation on mitochondrial function in insulin-resistant H9c2 cells. BMC Cardiovasc Disord 25(1):232. https://doi.org/10.1186/s12872-024-04397-7
-
Wang X, Copmans D, de Witte, et al (2021) Using zebrafish as a disease model to study fibrotic disease. Int J Mol Sci 22(12):6404. https://doi.org/10.3390/ijms22126404
-
Wei YL, Lin XC, Liu YY et al (2024) Effects of water fluoridation on early embryonic development of zebrafish. Ecotoxicol Environ Saf 270:115907. https://doi.org/10.1016/j.ecoenv.2023.115907
-
Xie J, Yan X, Xu G et al (2020) ITRAQ-based proteomics reveals the potential mechanism of fluoride-induced myocardial contraction function damage. Ecotoxicol Environ Saf 197:110605. https://doi.org/10.1016/j.ecoenv.2020.110605
-
Hou L, Dong H, Zhang E et al (2024) A new insight into fluoride induces cardiotoxicity in chickens: involving the regulation of PERK/IRE1/ATF6 pathway and heat shock proteins. Toxicology 501:153688. https://doi.org/10.1016/j.tox.2023.153688
-
Reddy YP, Tiwari S, Tomar LK et al (2021) Fluoride-induced expression of neuroinflammatory markers and neurophysiological regulation in the brain of Wistar rat model. Biol Trace Elem Res 199(7):2621–2626. https://doi.org/10.1007/s12011-020-02362-x
-
Kuang P, Cui H, Yu L (2022) Sodium fluoride suppresses spleen development through MAPK/ERK signaling pathway in mice. Ecotoxicol Environ Saf 241:113764. https://doi.org/10.1016/j.ecoenv.2022.113764
-
Chandimali N, Bak SG, Park EH et al (2025) Free radicals and their impact on health and antioxidant defenses: a review. Cell Death Discov 11:19. https://doi.org/10.1038/s41420-024-02278-8
-
Yan X, Feng C, Chen Q et al (2009) Effects of sodium fluoride treatment in vitro on cell proliferation, apoptosis and caspase-3 and caspase-9 mrna expression by neonatal rat osteoblasts. Arch Toxicol 83:451–458. https://doi.org/10.1007/s00204-008-0365-z
-
Ying J, Xu J, Shen L et al (2017) The effect of Sodium fluoride on cell apoptosis and the mechanism of human lung BEAS-2B cells in vitro. Biol Trace Elem Res 179(1):59–69. https://doi.org/10.1007/s12011-017-0937-y
-
Sahoo G, Samal D, Khandayataray P et al (2023) A review on caspases: key regulators of biological activities and apoptosis. Mol Neurobiol 60(10):5805–5837. https://doi.org/10.1007/s12035-023-03433-5
-
Zhang S, Yang Y, Lv X et al (2024) Doxorubicin-induced cardiotoxicity through SIRT1 loss potentiates overproduction of exosomes in cardiomyocytes. Int J Mol Sci 25(22):12376. https://doi.org/10.3390/ijms252212376
-
Chong ZZ, Wang S, Shang YC et al (2012) Targeting cardiovascular disease with novel SIRT1 pathways. Future Cardiol 8(1):89–100. https://doi.org/10.2217/fca.11.76
-
Giannakou ME, Partridge L (2004) The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol 14(8):408–412. https://doi.org/10.1016/j.tcb.2004.07.006
-
Yang Y, Liu Y, Wang Y et al (2022) Regulation of SIRT1 and its roles in inflammation. Front Immunol 13:831168. https://doi.org/10.3389/fimmu.2022.831168
-
Brunet A, Sweeney LB, Sturgill JF et al (2004) Stress-dependent regulation of FOXO transcription factors by SIRT1 deacetylase. Science 303(5666):2011–2015. https://doi.org/10.1126/science.1094637
-
Ding X, Zhu C, Wang W et al (2024) SIRT1 is a regulator of autophagy: implications for the progression and treatment of myocardial ischemia-reperfusion. Pharmacol Res 199:106957. https://doi.org/10.1016/j.phrs.2023.106957
-
Alcendor RR, Gao S, Zhai P et al (2007) Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res 100(10):1512–1521. https://doi.org/10.1161/01.RES.0000267723.65696.4a
-
Chen J, Niu Q, Xia T et al (2018) ERK1/2-mediated disruption of BDNF-TrkB signaling causes synaptic impairment contributing to fluoride-induced developmental neurotoxicity. Toxicology 410:222–230. https://doi.org/10.1016/j.tox.2018.08.009
-
Xu S, Yang Y, Han S et al (2014) ZIP1 and zinc inhibits fluoride-induced apoptosis in MC3T3-E1 cells. Biol Trace Elem Res 159(1–3):399–409. https://doi.org/10.1007/s12011-014-9935-5
Funding
This work was supported by the National Natural Science Foundation of China [Nos. 82274610, 81960818, 82060079]; The Natural Science Foundation of Guizhou Province (Nos. QianKeHe Support [2022]181, Qiankehe Cooperation Platform talents [2021] Postdoctoral Station 007, BSH[2024]005, ZK(2021)357); the Science and Technology Innovation Talent Team Project (Nos. GZYTD [2024] 003, GZYYFY2025002, GZYYFY2025010, GZSJDZX-2025-1-1,3,4,15, XueShuXinMiao[2023]07); the Research Project of Education Department of Guizhou Province (No. QianJiaoJi [2023]037). The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Ethics declarations
Consent to Participate
The full consent of all authors is confirmed.
Consent for Publication
All authors agree to submit this paper for publication.
Conflict of interest
The authors declare no competing interests.
FULL-TEXT STUDY ONLINE AT https://link.springer.com/article/10.1007/s12011-025-04828-2