Full Text:
As a strong oxidant, fluorine can induce oxidative stress resulting in cellular damage. Ferroptosis is an iron-dependent type of cell death caused by unrestricted lipid peroxidation (LPO) and subsequent plasma membrane rupture. This article indicated a relationship between fluorosis and ferroptosis. Evidence of the depletion of glutathione (GSH) and increased oxidized GSH can be found in a variety of organisms in high fluorine environments. Studies have shown that high fluoride levels can reduce the antioxidant capacity of antioxidant enzymes, while increasing the contents of reactive oxygen species (ROS) and malondialdehyde (MDA), resulting in oxidative stress and fluoride-induced oxidative stress, which are related to iron metabolism disorders. Excessive fluorine causes insufficient GSH, glutathione peroxidase (GSH-Px) inhibition, and oxidative stress, resulting in ferroptosis, which may play an important role in the occurrence and development of fluorosis.
Introduction
Fluorosis
Fluoride is widely present in nature and has strong oxidizing properties. Excessive fluoride in nature poses a serious threat to the health of plants, animals, and humans. Animal studies have shown that fluoride is neurotoxic, leading to learning and memory impairment, as well as teratogenic effects that can induce malformations and premature aging of the reproductive system (Perumal et al., 2013). Excessive fluoride intake during human growth and development can cause a variety of health problems from mild effects on teeth and bones to severe kidney problems, neurotoxicity, and even cancer (Saeed et al., 2020). Moreover, the fluoride content in drinking water has been positively correlated with tooth and bone fluorosis (Mondal et al., 2016). The “Guidelines for Drinking-Water Quality” state that a fluoride concentration in drinking water greater than 1.5 mg/L increases the risk of dental fluorosis (WHO, 2017).
During the development of tooth enamel, excessive fluoride can inhibit the activity of alkaline phosphatase and induce apoptosis of ameloblasts through oxidative stress (Yang et al., 2015), which adversely affects the normal mineralization process of tooth enamel, resulting in chalky or brown plaques on the enamel, and in severe cases, substantial enamel defects. Dental fluorosis is more prevalent in areas, where fluorine-rich coal is burned, drinking water is polluted, and brick tea is consumed. A recent meta-analysis showed that since the defluorination of drinking water in China (fluorine content from 2.72 to 0.54 mg/L), the prevalence and severity of fluorosis among children has decreased significantly over the past two decades (Wang et al., 2021). In addition to fluoride intake, the risk of disease is also closely related to the susceptibility of individual genes. Polymorphisms of the calcitonin receptor, type I collagen ?2 chain, and enamel matrix genes are thought to affect the occurrence of dental fluorosis (Jiang et al., 2015; Küchler et al., 2018).
Mechanisms of Fluorosis
Fluorine is a strong oxidant that can cause oxidative stress and subsequent cell damage. Studies have shown that within a certain period, the levels of intracellular reactive oxygen species (ROS) and apoptosis are linearly related to fluorine exposure (Ke et al., 2016). Fluoride causes lipid peroxidation (LPO), which destroys membrane phospholipids, resulting in extensive damage to intracellular organelles. In the endoplasmic reticulum of cells exposed to fluoride, the accumulation of unfolded and misfolded proteins in large quantities increases with fluoride concentrations, indicating that fluoride can induce the unfolded protein response and endoplasmic reticulum stress, which can trigger endoplasmic reticulum-mediated apoptosis (Liu et al., 2015).
Under physiological conditions, antioxidant cellular mechanisms regulate oxidative stress, but fluoride can inhibit gene expression and reduce the activity of superoxide dismutase (SOD; Wang et al., 2014). Due to the decreased expression and activity of antioxidant enzymes, the antioxidant capacity of cells is insufficient to resist the large amounts of ROS produced by fluoride, which results in cell damage, imbalance of cell redox homeostasis, and finally apoptosis.
Recent studies of the mechanisms underlying the onset of fluorosis have revealed that excessive fluorine can induce oxidative stress, regulate cellular redox homeostasis, and cause mitochondrial damage, endoplasmic reticulum stress, and altered gene expression (Zuo et al., 2018). This article proposes that ferroptosis may play a potential role in the occurrence and development of fluorosis, which can provide methods for the prevention and treatment of fluorosis.
Ferroptosis
Ferroptosis is an iron-dependent form of cell death caused by unrestricted LPO and subsequent plasma membrane rupture (Figure 1).
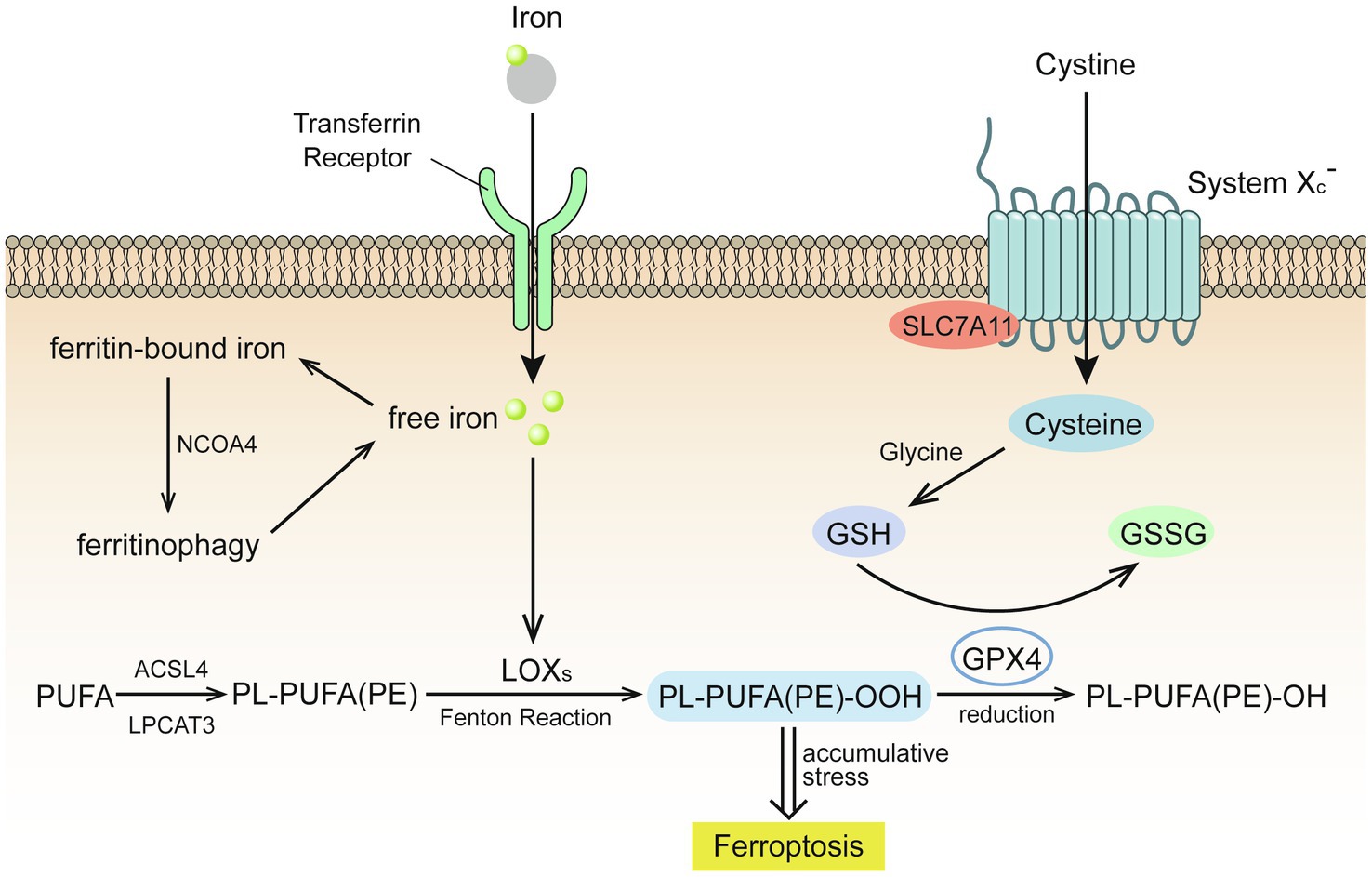 Figure 1. Glutathione (GSH) insufficient promotes the occurrence of ferroptosis (Stockwell et al., 2017). Extracellular cystine is exchanged with glutamate to enter the cell through the cystine/glutamate reverse transport system (system Xc-) and then reverts to cysteine. Under the catalysis of galactosidase and glutathione synthetase, cysteine and glycine co-synthesize glutathione (GSH), which is oxidized by glutathione peroxidase 4 (GPX4) to glutathione disulfide (GSSG), while GPX4 reduces toxic lipid peroxide to nontoxic lipid alcohol. Acyl-CoA synthetase long chain family member 4 (ACSL4) catalyzes the binding of free polyunsaturated fatty acids (PUFAs) to coenzyme A, and lysophosphatidylcholine acyltransferase 3 (LPCAT3) promotes their binding to membrane phospholipids to become PL-PUFAs (PE). Lipoxygenase (LOXs) mediates lipid peroxidation. PL-PUFA (PE) is oxidized to PL-PUFA (PE)-OOH, which becomes a ferroptosis signal. Extracellular iron ions enter the cell from the extracellular environment through transferrin and transferrin receptors, and generate reactive oxygen species (ROS) through the iron-dependent Fenton reaction or activate lipoxygenase to promote lipid peroxidation and promote ferroptosis.
Figure 1. Glutathione (GSH) insufficient promotes the occurrence of ferroptosis (Stockwell et al., 2017). Extracellular cystine is exchanged with glutamate to enter the cell through the cystine/glutamate reverse transport system (system Xc-) and then reverts to cysteine. Under the catalysis of galactosidase and glutathione synthetase, cysteine and glycine co-synthesize glutathione (GSH), which is oxidized by glutathione peroxidase 4 (GPX4) to glutathione disulfide (GSSG), while GPX4 reduces toxic lipid peroxide to nontoxic lipid alcohol. Acyl-CoA synthetase long chain family member 4 (ACSL4) catalyzes the binding of free polyunsaturated fatty acids (PUFAs) to coenzyme A, and lysophosphatidylcholine acyltransferase 3 (LPCAT3) promotes their binding to membrane phospholipids to become PL-PUFAs (PE). Lipoxygenase (LOXs) mediates lipid peroxidation. PL-PUFA (PE) is oxidized to PL-PUFA (PE)-OOH, which becomes a ferroptosis signal. Extracellular iron ions enter the cell from the extracellular environment through transferrin and transferrin receptors, and generate reactive oxygen species (ROS) through the iron-dependent Fenton reaction or activate lipoxygenase to promote lipid peroxidation and promote ferroptosis.
Amino Acid Metabolism
Extracellular cystine is exchanged with glutamate to enter the cell through the cystine/glutamate reverse transport system (system Xc-) and then reverts to cysteine. Under the catalysis of galactosidase and glutathione (GSH) synthetase, cysteine and glycine co-synthesize GSH, which is oxidized by glutathione peroxidase 4 (GPX4) to glutathione disulfide (Stockwell et al., 2017). GPX4 reduces toxic lipid peroxide (L-OOH) to nontoxic lipid alcohol (L-OH), which requires the catalytic selenocysteine residue of GPX4 and two electrons provided mainly by GSH (Jiang et al., 2021).
The lack of GSH leads to GPX4 inhibition, which causes cell damage due to excessive lipid peroxides, results in cell ferroptosis (Stockwell et al., 2017). The expression and activity of solute carrier family 7 member 11 and solute carrier family 3 member 2, two subunits of system Xc-, control the transport of cystine (Jiang et al., 2021). Excessive concentrations of extracellular glutamate will inhibit the transport of cystine, resulting in insufficient GSH synthesis.
Lipid Metabolism
Acyl-CoA synthetase long chain family member 4 (ACSL4) catalyzes the binding of free polyunsaturated fatty acids (PUFAs), such as arachidonic acid and adrenal acid, to coenzyme A, and lysophosphatidylcholine acyltransferase 3 (LPCAT3) promotes their binding to membrane phospholipids to become PL-PUFAs (PE). Arachidonate lipoxygenase (ALOX) mediates lipid peroxidation. Membrane electron transfer proteins participate in the production of ROS during lipid peroxidation. PL-PUFA (PE) is oxidized to PL-PUFA (PE)-OOH, which becomes a ferroptosis signal (Jiang et al., 2021). Reduced production of AA-PE membrane phospholipids inhibits lipid peroxidation and ferroptosis. For example, ACSL3 binds monounsaturated fatty acids to membrane phospholipids and 5′ AMP-activated protein kinase (AMPK)-mediated phosphorylation of acetyl-CoA carboxylase alpha limits the production of PUFAs. Conversely, increasing the production of AA-PE membrane phospholipids promotes lipid peroxidation and ferroptosis. For example, as the activities of ACSL4, LPCAT3, and ALOX increase, the intake of PUFAs also increases, while AMPK-mediated phosphorylation of beclin1 inhibits GSH production (Chen et al., 2021).
Iron Metabolism
Extracellular iron ions enter the cell from the extracellular environment through transferrin and transferrin receptors. Upon recognition by nuclear receptor coactivator 4, intracellular ferritin is autophagocytosed and degraded by lysosomes to release free iron and generate ROS through the iron-dependent Fenton reaction or activate lipoxygenase to promote lipid peroxidation and promote ferroptosis. The Fenton reaction is a process by which excessive iron ions and hydrogen peroxide in the cell generate highly toxic hydroxyl radicals (Qian et al., 2019), which are than combined with the double bond of PL-PUFA to generate lipid peroxide (Tkachenko et al., 2021).
Drugs that increase iron-mediated toxicity, transferrin, and ferritin autophagy promote ferroptosis by increasing the intracellular content of free iron, while iron-chelating drugs, solute carrier family 40 member 1, exosomal-mediated ferritin export, mitochondrial protein-mediated iron-sulfur group biogenesis, and silencing of iron responsive element binding protein 2 have an opposite effect (Jiang et al., 2021).
Relationship Between Fluorosis and Ferroptosis
Fluoride-resistant MC3T3-E1 (FR MC3T3-E1) cells, were inducted by sub-cultivating MC3T3-E1 cells (WT MC3T3-E1) in fluoride medium of 10 ppm fluoride concentration gradient ascending media, as mentioned in the previous article (Ran et al., 2017; Figure 2A). In order to explore the changes of ferroptosis-related gene expression in 30 ppm FR MC3T3-E1 cells, we performed real-time PCR (Figure 2B). The expression levels of these genes suggest that in the fluoride-containing medium, the ferroptosis degree in FR cells is lower than that in WT cells, indicating that there is a potential connection between cell fluorosis and ferroptosis. Although direct proof that excessive fluorine causes cell ferroptosis is lacking, the following studies can infer the relationship between fluorosis and ferroptosis.
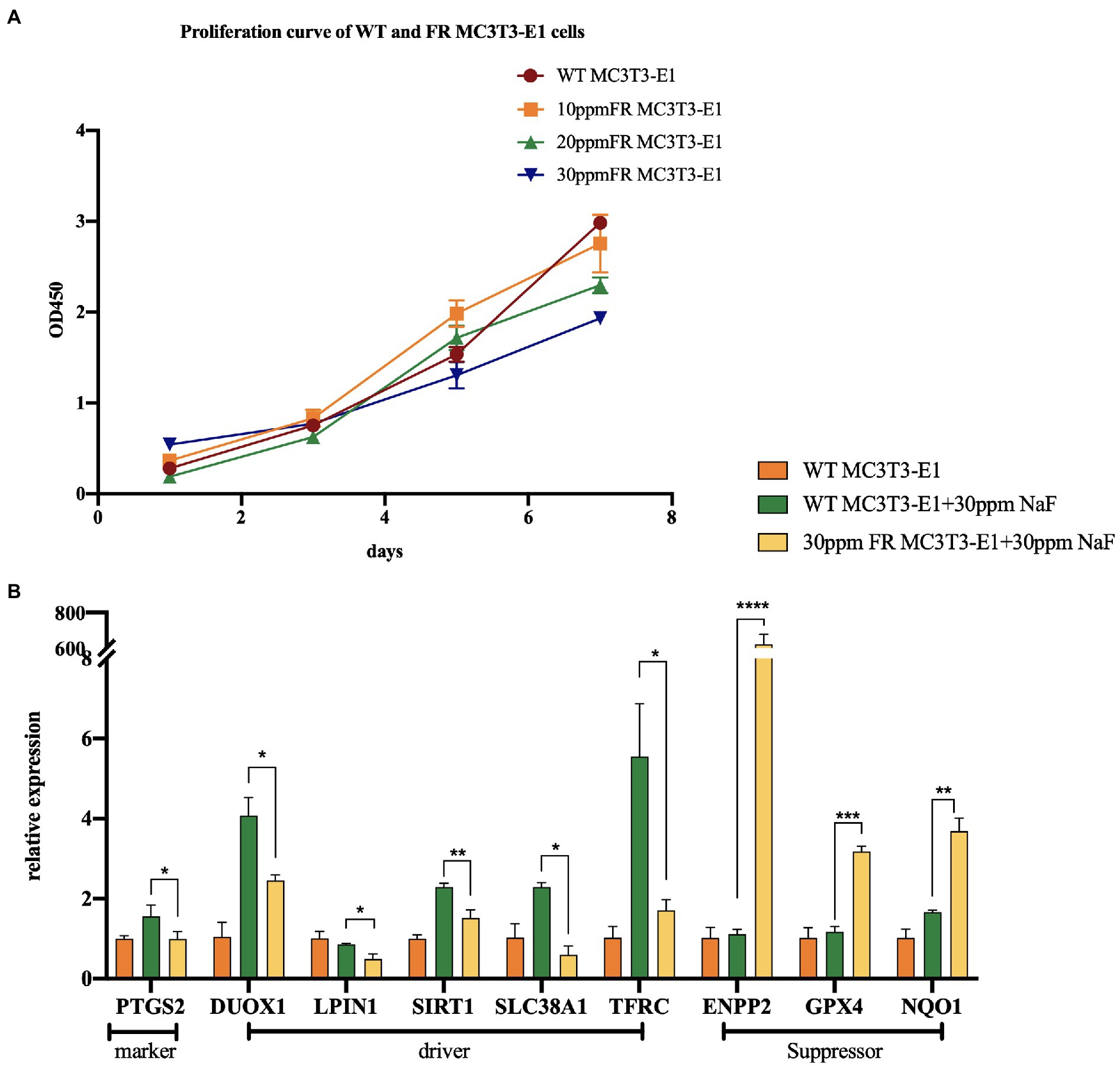 Figure 2. The changes of ferroptosis-related gene expression in fluoride-resistant MC3T3-E1 cells. (A) Cell-Counting-Kit-8 assay was used to determine the cell proliferation rate. Fluoride-resistant MC3T3-E1 (FR MC3T3-E1) cells, were inducted by sub-cultivating in 10 ppm fluoride concentration gradient ascending media, and continued till the proliferation rate of the cells in the fluoride media became normal. (B) Real-time PCR showed the changes of ferroptosis-related gene expression between 30 ppm FR MC3T3-E1 cells and WT MC3T3-E1 cells. When cultured in 30 ppm fluoride medium, the expression of Prostaglandin-Endoperoxide Synthase 2 (PTGS2) in FR cells was lower than that in WT cells, which is the marker of ferroptosis. Dual Oxidase 1 (DUOX1), Lipin 1 (LIPIN1), Sirtuin 1 (SIRT1), Solute Carrier Family 38 Member 1 (SCL38A1), and Transferrin Receptor (TFRC) are the drivers of ferroptosis. Ectonucleotide Pyrophosphatase/Phosphodiesterase 2 (ENPP2), GPX4, NAD(P)H Quinone Dehydrogenase 1 (NQO1) are ferroptosis suppressors. The result indicates that the expression of ferroptosis driver in FR cells is lower than that in WT cells. Similarly, the expression of ferroptosis suppressor in FR cells was significantly higher than that in WT cells. *p < 0.05; **p < 0.01; ***p < 0.001 and ****p < 0.0001.
Figure 2. The changes of ferroptosis-related gene expression in fluoride-resistant MC3T3-E1 cells. (A) Cell-Counting-Kit-8 assay was used to determine the cell proliferation rate. Fluoride-resistant MC3T3-E1 (FR MC3T3-E1) cells, were inducted by sub-cultivating in 10 ppm fluoride concentration gradient ascending media, and continued till the proliferation rate of the cells in the fluoride media became normal. (B) Real-time PCR showed the changes of ferroptosis-related gene expression between 30 ppm FR MC3T3-E1 cells and WT MC3T3-E1 cells. When cultured in 30 ppm fluoride medium, the expression of Prostaglandin-Endoperoxide Synthase 2 (PTGS2) in FR cells was lower than that in WT cells, which is the marker of ferroptosis. Dual Oxidase 1 (DUOX1), Lipin 1 (LIPIN1), Sirtuin 1 (SIRT1), Solute Carrier Family 38 Member 1 (SCL38A1), and Transferrin Receptor (TFRC) are the drivers of ferroptosis. Ectonucleotide Pyrophosphatase/Phosphodiesterase 2 (ENPP2), GPX4, NAD(P)H Quinone Dehydrogenase 1 (NQO1) are ferroptosis suppressors. The result indicates that the expression of ferroptosis driver in FR cells is lower than that in WT cells. Similarly, the expression of ferroptosis suppressor in FR cells was significantly higher than that in WT cells. *p < 0.05; **p < 0.01; ***p < 0.001 and ****p < 0.0001.
Fluorosis in a Variety of Organisms Shows Glutathione and Glutathione Peroxidase Depletion
Evidence of the depletion of GSH can be found in a variety of organisms in high fluorine environments, which is oxidized by glutathione peroxidase 4 (GPX4) to glutathione disulfide. As a result of GPX4 reduces L-OOH to L-OH (Stockwell et al., 2017), insufficient GSH is reported to inhibit the ability of GPX4 to reduce lipid peroxides, thereby promoting the occurrence of ferroptosis (Figure 1).
Perfluorooctane sulfonyl fluoride (PFOSF) is the precursor of many fluorine-containing compounds that are ubiquitous in the environment. Preliminary toxicological studies have confirmed that PFOSF can promote the oxidation of GSH to glutathione disulfide, which is then reduced to sulfinic acid (Jin et al., 2020).
Digital gene expression, gene ontology, and the Kyoto Encyclopedia of Genes and Genomes analysis of the transcriptome profile of the silkworm testis treated with NaF showed that many differentially expressed genes are involved in the stress response and oxidative phosphorylation. In the NaF treatment group, the activity of glutathione S-transferase increased, while the content of GSH decreased (Tang et al., 2018). At a concentration of 10 mg/L in water, fluoride can induce antioxidant responses of aquatic plants, while excessive fluoride (20–40 mg/L) results in unbalanced metabolism and oxidative damage (Gao et al., 2018). Excessive fluoride concentrations in water have detrimental effects on liver and kidney function, and ovarian development in fish, while reducing the production of reproductive hormones and eggs. There is a positive correlation between the hepatocyte apoptosis index and fluoride levels. Fluoride-induced oxidative stress in fish manifests as an increase in lipid peroxidation and consumption of GSH levels. The activity and mRNA expression levels of antioxidant enzymes, such as SOD and glutathione peroxidase (GSH-Px), significantly decreased, while the content of malondialdehyde (MDA), the end product of lipid peroxidation, significantly increased (Cao et al., 2013, 2015; Li et al., 2020).
Similar conclusions have been obtained in mammals. Excessive fluoride intake can cause oxidative stress and damage the antioxidant system in chickens. It reduced the activities of SOD and GSH-Px, increased ROS and MDA (Chen et al., 2011; Miao et al., 2017; Wang et al., 2018). Research on the antioxidant status of buffalo calves found that NaF exposure led to increased oxidative stress and a significant increase in LPO with significant reductions in GSH-Px and catalase (CAT) activities and GSH levels (Gill and Dumka, 2016).
Excessive sodium fluoride can cause pathological changes in the morphologies of the rat liver and lung, as well as increase serum levels of aspartate aminotransferase, alanine aminotransferase, and MDA, while decreasing total antioxidant capacity (T-AOC) and serum levels of SOD and GSH-Px (Mohammed et al., 2017; Mittal et al., 2018; Li et al., 2021). It has been reported that exposure to 10 ppm of fluoride will significantly increase serum level of ROS in rats, while reducing serum levels of GSH. Interestingly, exposure to fluorine at 50 and 100 ppm will not produce more obvious toxicity, thus the underlying mechanism of action is unclear (Chouhan and Flora, 2008). Similarly, excessive intake of fluoride can reduce the developmental potential of mouse oocytes by inducing oxidative stress and apoptosis. The depletion of antioxidant enzymes, such as GSH, glucose 6 phosphate dehydrogenase, SOD, and GSH-Px, with an increase in nitric oxide synthase 2 was detected in the mouse liver (Zhou et al., 2015; Wang et al., 2017). As compared to the brain, the liver is more sensitive to the toxic effects of fluoride. Interestingly, the combined exposure of arsenic and fluoride causes less obvious toxicity, and the joint exposure of these two toxic substances does not cause synergistic toxicity. In some cases, joint exposure to arsenic and fluoride will have an antagonistic effect (Mittal and Flora, 2006). Cytology experiments have found that NaF at concentrations of 5–20 mg/L may cause central nervous system poisoning by stimulating the conversion of BV-2 (mouse microglia) cells into activated microglia. In BV-2 cells treated with fluoride, SOD levels decreased, while those of MDA, ROS, superoxide anions (O2?), and NOS significantly increased in a dose-dependent manner (Shuhua et al., 2012).
It is found that in southwestern China, the people with endemic fluorosis had significantly reduced the levels of antioxidant enzyme activity including copper/zinc SOD, GSH-Px, and CAT, while those exposed to higher levels of fluorine had significantly lower serum antioxidant enzyme activities (Wang et al., 2014). Cytological studies found that exposure to 0.1 and 1% ammonium hexafluorosilicate (SiF) for 10 and 30 min caused a sharp increase in the cellular GSH consumption of human gingival fibroblasts (Song et al., 2013). Studies on human hepatocytes (L-02) have found that fluoride causes an increase in the LPO content of L-02 cells and a decrease in GSH content, with a significant dose-effect relationship (Wang et al., 2004).
Fluorosis and Lipid Peroxidation
Aldehydes, such as MDA, as the final product of lipid peroxidation, produce toxicity by changing the structure and function of nucleic acids and proteins, and thus are considered as the “execution” stage of ferroptosis (Gaschler and Stockwell, 2017). The level of Thiobarbituric Acid Reactive Substances (TBARS) test represents the level of MDA, which is used to measure the levels of lipid peroxidation. The TBARS levels in the kidneys, lungs, and testes of rats exposed to 30 mg/L of NaF throughout pregnancy and lactation were higher than those in the control group, and significant tissue destruction was observed (Karaoz et al., 2004; Oncu et al., 2006, 2007). The TBARS levels in blood samples of Ukrainian children with chronic fluorosis (exposed to drinking water fluoride at >1.5 ppm for >5 years) in endemic areas were higher than those of healthy children living in non-fluorosis areas, indicating that fluorosis increased lipid peroxidation (Tkachenko et al., 2021).
Dietary fluoride supplementation leads to decrease antioxidant enzyme activities in the pig liver, in addition to decreased abilities to scavenge free radicals and excessive production of LPO (Zhan et al., 2006). Mice exposed to fluoride (NaF) at 15 ppm for 8 months showed increased consumption of GSH, lipid peroxidation, and catalase activity in the liver and brain. Fluorine-induced DNA damage and damaged DNA repair mechanisms trigger liver and brain cell apoptosis and tissue structure damage (Bhowmik et al., 2020). Therefore, high fluoride levels can reduce the activities of antioxidant enzymes, including SOD, CAT and GSH-Px, while simultaneously increasing ROS and MDA contents, resulting in oxidative stress.
Fluorosis and Iron Metabolism
Extracellular iron ions enter the cell from the extracellular environment through transferrin and transferrin receptors. Upon recognition by nuclear receptor coactivator 4, intracellular ferritin is autophagocytosed and degraded by lysosomes to release free iron and generate ROS through the iron-dependent Fenton reaction or activate lipoxygenase to promote lipid peroxidation and promote ferroptosis (D’Herde and Krysko, 2017).
Fluorosis can significantly change mineral metabolism. For example, the kidney tissue of sheep in areas with endemic fluorosis reportedly had significantly higher iron concentrations (Çetin and Yur, 2016). Fluoride-induced oxidative stress has been correlated to iron metabolism disorders, which are characterized by increased iron and hepcidin levels, but decreased expression of iron transporters (Niu et al., 2018). In vitro experiments found that human red blood cells treated with NaF produced methemoglobin and oxyhemoglobin, with increased heme degradation, resulting in the release of free iron from porphyrin rings (Maheshwari et al., 2021). These findings suggested that fluoride exposure may have a potential impact on iron metabolism.
Perspectives and Conclusion
It is generally believed that the mechanism of fluorosis mainly revolves around oxidative stress, enzyme and protein inactivation, acid-base balance, and electrolyte metabolism disorders. However, excessive fluorine may cause a decrease in GSH with a simultaneous decrease the activity of antioxidant enzymes, such as SOD and GSH-Px, while the content of MDA, the end product of lipid peroxidation, significantly increase. They all indicate an increase in ferroptosis. Our study result indicated that ferroptosis may play an important role in the occurrence of fluorosis. However, it just a preliminary experiment based in vitro. Further researches are needed to determine the role of ferroptosis in fluorosis. Secondly, it is necessary to determine the pathway of ferroptosis participate in the occurrence and development of fluorosis, and then to determine the relationship between fluoride and these pathways. These studies will provide solutions for the prevention and treatment of fluorosis.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
YZ, JW, LJ, and CL collected and analyzed the data. YZ drafted the article. BL and ZH revised the article. All authors contributed to the article and approved the submitted version.
Funding
This work was sponsored by Natural Science Foundation of Shanghai (grant No. 21ZR1455700) and the Research Discipline fund (No. KQYJXK2020) from Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, and College of Stomatology, Shanghai Jiao Tong University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank YM Fang for amendments to the article and Shen Zhao performed the revising and final approval in major revision.
References
Bhowmik, A. D., Podder, S., Mondal, P., Shaw, P., Bandyopadhyay, A., Das, A., et al. (2020). Chronic exposure to environmentally relevant concentration of fluoride alters Ogg1 and Rad51 expressions in mice: involvement of epigenetic regulation. Ecotoxicol. Environ. Saf. 202:110962. doi: 10.1016/j.ecoenv.2020.110962
Cao, J., Chen, J., Wang, J., Jia, R., Xue, W., Luo, Y., et al. (2013). Effects of fluoride on liver apoptosis and Bcl-2, Bax protein expression in freshwater teleost, Cyprinus carpio. Chemosphere 91, 1203–1212. doi: 10.1016/j.chemosphere.2013.01.037
Cao, J., Chen, J., Xie, L., Wang, J., Feng, C., and Song, J. (2015). Protective properties of sesamin against fluoride-induced oxidative stress and apoptosis in kidney of carp (Cyprinus carpio) via JNK signaling pathway. Aquat. Toxicol. 167, 180–190. doi: 10.1016/j.aquatox.2015.08.004
Çetin, S., and Yur, F. (2016). Levels of trace elements in muscle and kidney tissues of sheep with fluorosis. Biol. Trace Elem. Res. 174, 82–84. doi: 10.1007/s12011-016-0694-3
Chen, T., Cui, H., Cui, Y., Bai, C., and Gong, T. (2011). Decreased antioxidase activities and oxidative stress in the spleen of chickens fed on high-fluorine diets. Hum. Exp. Toxicol. 30, 1282–1286. doi: 10.1177/0960327110388538
Chen, X., Kang, R., Kroemer, G., and Tang, D. (2021). Broadening horizons: the role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 18, 280–296. doi: 10.1038/s41571-020-00462-0
Chouhan, S., and Flora, S. J. (2008). Effects of fluoride on the tissue oxidative stress and apoptosis in rats: biochemical assays supported by IR spectroscopy data. Toxicology 254, 61–67. doi: 10.1016/j.tox.2008.09.008
D’Herde, K., and Krysko, D. V. (2017). Ferroptosis: oxidized PEs trigger death. Nat. Chem. Biol. 13, 4–5. doi: 10.1038/nchembio.2261
Gao, J., Liu, C., Zhang, J., Zhu, S., Shen, Y., and Zhang, R. (2018). Effect of fluoride on photosynthetic pigment content and antioxidant system of Hydrilla verticillata. Int. J. Phytoremediation 20, 1257–1263. doi: 10.1080/15226514.2017.1319328
Gaschler, M. M., and Stockwell, B. R. (2017). Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 482, 419–425. doi: 10.1016/j.bbrc.2016.10.086
Gill, K. K., and Dumka, V. K. (2016). Antioxidant status in oral subchronic toxicity of fipronil and fluoride co-exposure in buffalo calves. Toxicol. Ind. Health 32, 251–259. doi: 10.1177/0748233713500376
Jiang, M., Mu, L., Wang, Y., Yan, W., and Jiao, Y. (2015). The relationship between Alu I polymorphisms in the calcitonin receptor gene and fluorosis endemic to Chongqing, China. Med. Princ. Pract. 24, 80–83. doi: 10.1159/000368435
Jiang, X., Stockwell, B. R., and Conrad, M. (2021). Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282. doi: 10.1038/s41580-020-00324-8
Jin, Z., He, Q., Luo, H., Pan, Y., Sun, C., and Cai, Z. (2020). The oxidation of cysteine-containing peptides caused by perfluoroalkane sulfonyl fluorides. J. Hazard. Mater. 385:121564. doi: 10.1016/j.jhazmat.2019.121564
Karaoz, E., Oncu, M., Gulle, K., Kanter, M., Gultekin, F., Karaoz, S., et al. (2004). Effect of chronic fluorosis on lipid peroxidation and histology of kidney tissues in first- and second-generation rats. Biol. Trace Elem. Res. 102, 199–208. doi: 10.1385/BTER:102:1-3:199
Ke, L., Zheng, X., Sun, Y., Ouyang, W., and Zhang, Z. (2016). Effects of sodium fluoride on lipid peroxidation and PARP, XBP-1 expression in PC12 cell. Biol. Trace Elem. Res. 173, 161–167. doi: 10.1007/s12011-016-0641-3
Küchler, E. C., Bruzamolin, C. D., Omori, M. A., Costa, M. C., Antunes, L. S., Pecharki, G. D., et al. (2018). Polymorphisms in nonamelogenin enamel matrix genes are associated with dental fluorosis. Caries Res. 52, 1–6. doi: 10.1159/000479826
Li, M., Cao, J., Zhao, Y., Wu, P., Li, X., Khodaei, F., et al. (2020). Fluoride impairs ovary development by affecting oogenesis and inducing oxidative stress and apoptosis in female zebrafish (Danio rerio). Chemosphere 256:127105. doi: 10.1016/j.chemosphere.2020.127105
Li, H., Hao, Z., Wang, L., Yang, J., Zhao, Y., Cheng, X., et al. (2021). Dietary calcium alleviates fluorine-induced liver injury in rats by mitochondrial apoptosis pathway. Biol. Trace Elem. Res. 200, 271–280. doi: 10.1007/s12011-021-02641-1
Liu, L., Zhang, Y., Gu, H., Zhang, K., and Ma, L. (2015). Fluorosis induces endoplasmic reticulum stress and apoptosis in osteoblasts in vivo. Biol. Trace Elem. Res. 164, 64–71. doi: 10.1007/s12011-014-0192-4
Maheshwari, N., Qasim, N., Anjum, R., and Mahmood, R. (2021). Fluoride enhances generation of reactive oxygen and nitrogen species, oxidizes hemoglobin, lowers antioxidant power and inhibits transmembrane electron transport in isolated human red blood cells. Ecotoxicol. Environ. Saf. 208:111611. doi: 10.1016/j.ecoenv.2020.111611
Miao, L. P., Zhou, M. Y., Zhang, X. Y., Yuan, C., Dong, X. Y., and Zou, X. T. (2017). Effect of excess dietary fluoride on laying performance and antioxidant capacity of laying hens. Poult. Sci. 96, 2200–2205. doi: 10.3382/ps/pex0002
Mittal, M., Chatterjee, S., and Flora, S. J. S. (2018). Combination therapy with vitamin C and DMSA for arsenic-fluoride co-exposure in rats. Metallomics 10, 1291–1306. doi: 10.1039/C8MT00192H
Mittal, M., and Flora, S. J. (2006). Effects of individual and combined exposure to sodium arsenite and sodium fluoride on tissue oxidative stress, arsenic and fluoride levels in male mice. Chem. Biol. Interact. 162, 128–139. doi: 10.1016/j.cbi.2006.05.018
Mohammed, A. T., Mohamed, A. A., and Ali, H. (2017). Pulmonary apoptotic and oxidative damaging effects of Triclosan alone or in combination with fluoride in Sprague Dawley rats. Acta Histochem. 119, 357–363. doi: 10.1016/j.acthis.2017.03.004
Mondal, D., Dutta, G., and Gupta, S. (2016). Inferring the fluoride hydrogeochemistry and effect of consuming fluoride-contaminated drinking water on human health in some endemic areas of Birbhum district, West Bengal. Environ. Geochem. Health 38, 557–576. doi: 10.1007/s10653-015-9743-7
Niu, Q., He, P., Xu, S., Ma, R., Ding, Y., Mu, L., et al. (2018). Fluoride-induced iron overload contributes to hepatic oxidative damage in mouse and the protective role of grape seed proanthocyanidin extract. J. Toxicol. Sci. 43, 311–319. doi: 10.2131/jts.43.311
Oncu, M., Gulle, K., Karaoz, E., Gultekin, F., Karaoz, S., Karakoyun, I., et al. (2006). Effect of chronic fluorosis on lipid peroxidation and histology of lung tissues in first and second generation rats. Toxicol. Ind. Health 22, 375–380. doi: 10.1177/0748233706071973
Oncü, M., Kocak, A., Karaoz, E., Darici, H., Savik, E., and Gultekin, F. (2007). Effect of long-term fluoride exposure on lipid peroxidation and histology of testes in first- and second-generation rats. Biol. Trace Elem. Res. 118, 260–268. doi: 10.1007/s12011-007-0036-6
Perumal, E., Paul, V., Govindarajan, V., and Panneerselvam, L. (2013). A brief review on experimental fluorosis. Toxicol. Lett. 223, 236–251. doi: 10.1016/j.toxlet.2013.09.005
Qian, X., Zhang, J., Gu, Z., and Chen, Y. (2019). Nanocatalysts-augmented Fenton chemical reaction for nanocatalytic tumor therapy. Biomaterials 211, 1–13. doi: 10.1016/j.biomaterials.2019.04.023
Ran, S., Sun, N., Liu, Y., Zhang, W., Li, Y., Wei, L., et al. (2017). Fluoride resistance capacity in mammalian cells involves complex global gene expression changes. FEBS Open Bio 7, 968–980. doi: 10.1002/2211-5463.12236
Saeed, M., Malik, R. N., and Kamal, A. (2020). Fluorosis and cognitive development among children (6-14 years of age) in the endemic areas of the world: a review and critical analysis. Environ. Sci. Pollut. Res. Int. 27, 2566–2579. doi: 10.1007/s11356-019-06938-6
Shuhua, X., Ziyou, L., Ling, Y., Fei, W., and Sun, G. (2012). A role of fluoride on free radical generation and oxidative stress in BV-2 microglia cells. Mediat. Inflamm. 2012:102954. doi: 10.1155/2012/102954
Song, D. X., Zheng, L. W., Shen, S. M., and Chen, X. M. (2013). Cytotoxicity of ammonium hexafluorosilicate on human gingival fibroblasts. Toxicol. in Vitro 27, 2149–2155. doi: 10.1016/j.tiv.2013.09.005
Stockwell, B. R., Angeli, J. P. F., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Tang, W., Xiao, Y., Li, G., Zheng, X., Yin, Y., Wang, L., et al. (2018). Analysis of digital gene expression profiling in the gonad of male silkworms (Bombyx mori) under fluoride stress. Ecotoxicol. Environ. Saf. 153, 127–134. doi: 10.1016/j.ecoenv.2018.01.028
Tkachenko, H., Kurhaluk, N., Skaletska, N., Maksin, V., and Osadowski, Z. (2021). Elemental status and lipid peroxidation in the blood of children with endemic fluorosis. Biol. Trace Elem. Res. 199, 1237–1245. doi: 10.1007/s12011-020-02243-3
Wang, Q., Cui, K. P., Xu, Y. Y., Gao, Y. L., Zhao, J., Li, D. S., et al. (2014). Coal-burning endemic fluorosis is associated with reduced activity in antioxidative enzymes and cu/Zn-SOD gene expression. Environ. Geochem. Health 36, 107–115. doi: 10.1007/s10653-013-9522-2
Wang, F., Li, Y., Tang, D., Zhao, J., Yang, X., Liu, Y., et al. (2021). Effects of water improvement and defluoridation on fluorosis-endemic areas in China: a meta-analysis. Environ. Pollut. 270:116227. doi: 10.1016/j.envpol.2020.116227
Wang, A. G., Xia, T., Chu, Q. L., Zhang, M., Liu, F., Chen, X. M., et al. (2004). Effects of fluoride on lipid peroxidation, DNA damage and apoptosis in human embryo hepatocytes. Biomed. Environ. Sci. 17, 217–222.
Wang, Y. X., Xiao, X., and Zhan, X. A. (2018). Antagonistic effects of different selenium sources on growth inhibition, oxidative damage, and apoptosis induced by fluorine in broilers. Poult. Sci. 97, 3207–3217. doi: 10.3382/ps/pey192
Wang, H. W., Zhao, W. P., Liu, J., Tan, P. P., Zhang, C., and Zhou, B. H. (2017). Fluoride-induced oxidative stress and apoptosis are involved in the reducing of oocytes development potential in mice. Chemosphere 186, 911–918. doi: 10.1016/j.chemosphere.2017.08.068
WHO (2017). WHO Guidelines Approved by the Guidelines Review Committee. In Guidelines for Drinking-Water Quality: Fourth Edition Incorporating the First Addendum. Geneva: World Health Organization Copyright.
Yang, T., Zhang, Y., Zheng, D., Hao, Y., Snead, M. L., and Duan, X. (2015). High-fluoride promoted phagocytosis-induced apoptosis in a matured ameloblast-like cell line. Arch. Oral Biol. 60, 84–90. doi: 10.1016/j.archoralbio.2014.09.001
Zhan, X. A., Wang, M., Xu, Z. R., Li, W. F., and Li, J. X. (2006). Effects of fluoride on hepatic antioxidant system and transcription of cu/Zn SOD gene in young pigs. J. Trace Elem. Med. Biol. 20, 83–87. doi: 10.1016/j.jtemb.2005.11.003
Zhou, B. H., Zhao, J., Liu, J., Zhang, J. L., Li, J., and Wang, H. W. (2015). Fluoride-induced oxidative stress is involved in the morphological damage and dysfunction of liver in female mice. Chemosphere 139, 504–511. doi: 10.1016/j.chemosphere.2015.08.030
Keywords: fluorosis, ferroptosis, oxidative stress, antioxidant enzymes, lipid peroxidation, iron metabolism
Citation: Zhang Y, Wu J, Jiang L, Lu C, Huang Z and Liu B (2021) Prospects for the Role of Ferroptosis in Fluorosis. Front. Physiol. 12:773055. doi: 10.3389/fphys.2021.773055
Edited by:
Jorge E. Araya, University of Antofagasta, Chile
Reviewed by:
Mauricio Zamorano, University of La Frontera, Chile
Zhili Li, China Astronaut Research and Training Center, China
Copyright © 2021 Zhang, Wu, Jiang, Lu, Huang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Liu, pizzanet@163.com; Zhengwei Huang, huangzhengwei@shsmu.edu.cn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
*Original study online at https://www.frontiersin.org/articles/10.3389/fphys.2021.773055/full